

Introduction
Sodium (Na⁺) channels are integral membrane proteins responsible for the rapid upstroke (phase 0) of the action potential in excitable cells such as neurons and cardiomyocytes. By selectively perming sodium ions across membranes, these channels facilitate depolarization, triggering electrical signals essential for neurotransmission, skeletal muscle contraction, and cardiac conduction. As a result, therapeutic modulation of sodium channels can manage a variety of pathophysiological conditions, from arrhythmias to chronic pain and epilepsy.
- Introduction
- Sodium Channel Physiology and Structure
- Classification of Sodium Channel Blockers
- Mechanism of Action and Kinetics
- Clinical Applications of Sodium Channel Blockers
- Adverse Effects and Toxicity
- Pharmacokinetic Considerations
- Clinical Pearls for Use
- Emerging and Investigational Sodium Channel Blockers
- Special Populations
- Clinical Vignette Examples
- Future Directions and Summary
- References
Drugs collectively known as sodium channel blockers exert their effects by binding to and stabilizing certain conformational states of the channel, reducing sodium influx and dampening cellular excitability. This broad class includes antiarrhythmic drugs (particularly Class I agents), local anesthetics, and many anticonvulsants. Though sharing similar foundational mechanisms, these agents differ in their kinetics, state-dependent binding, tissue selectivity, and clinical indications.
This article provides a comprehensive review of sodium channel blockers, summarizing their classification, molecular dynamics, clinical applications, and adverse effects. By understanding how these agents manipulate sodium channels, clinicians and researchers can more precisely tailor drug therapy to individual pathologies, aiming to minimize toxicities while maximizing therapeutic benefit.
Sodium Channel Physiology and Structure
Sodium Channel Subtypes
Voltage-gated sodium (Naᵥ) channels are large transmembrane complexes composed of an alpha subunit and one or more auxiliary beta subunits. The alpha subunit—responsible for ion conduction and voltage sensing—contains four homologous domains (I–IV), each featuring six transmembrane segments (S1–S6). Variations in alpha subunit isoforms yield multiple Naᵥ subtypes, expressed throughout the body (e.g., Naᵥ1.1, Naᵥ1.2, Naᵥ1.4, Naᵥ1.5, etc.).For example, Naᵥ1.5 is abundantly found in cardiac tissue, pivotal for the fast sodium current underlying cardiac action potentials. Naᵥ1.7, Naᵥ1.8, and Naᵥ1.9 are more common in peripheral nerves, linked to pain pathways. Meanwhile, Naᵥ1.2 and Naᵥ1.6 predominate in the central nervous system, integral to neuronal firing. Understanding these isoforms helps explain why certain sodium channel blockers selectively target the heart or nerves.
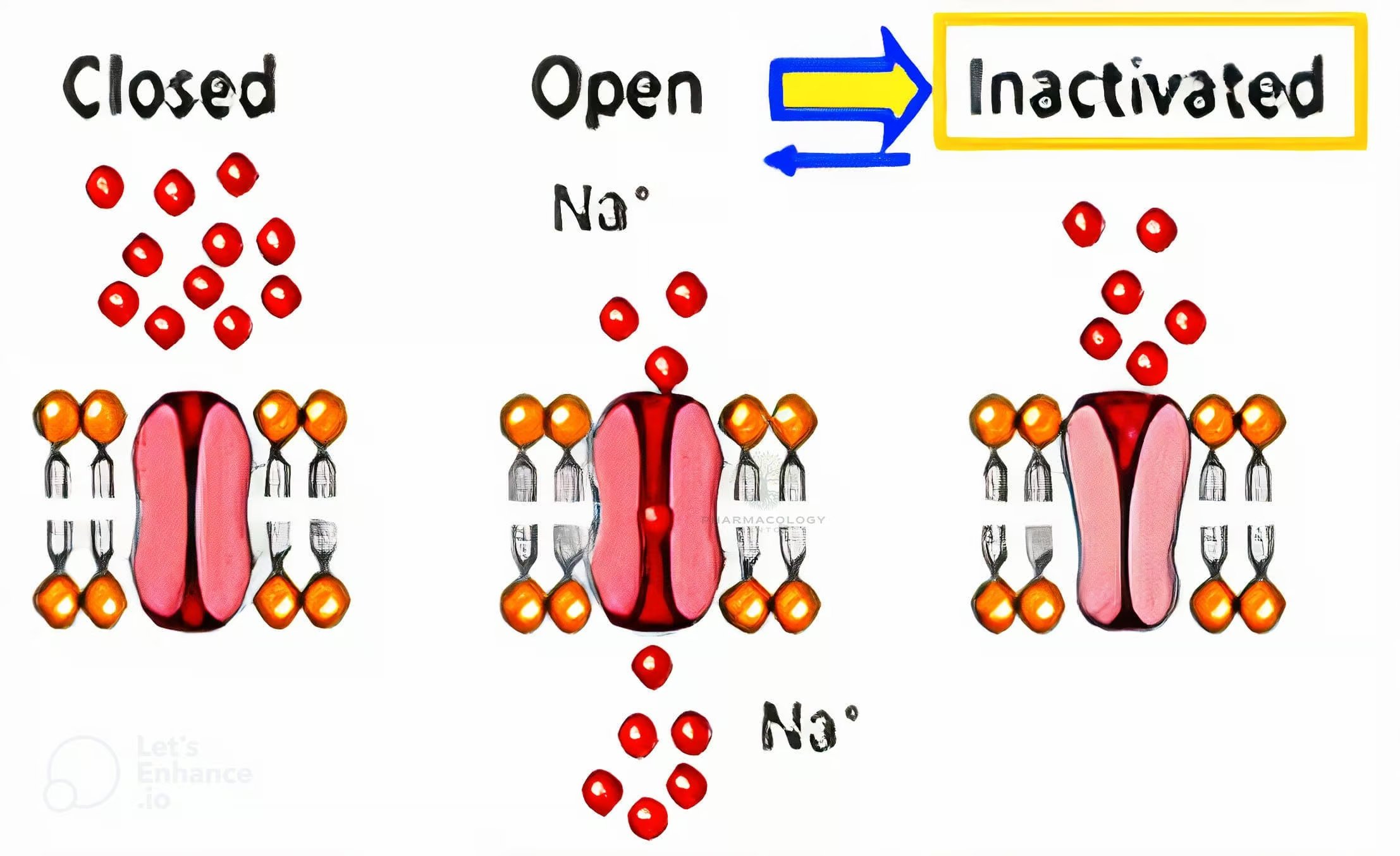
Channel Conformational States
During an action potential, sodium channels transition through three principle states:
- Resting (closed): The channel is closed but can open if the membrane depolarizes.
- Open (activated): Rapid Na⁺ influx depolarizes the cell, generating an action potential upstroke.
- Inactivated (closed): The channel is nonconducting, incapable of immediate reopening until repolarization.
Many sodium channel blockers exhibit state-dependent binding, meaning they have higher affinity for channels in the activated or inactivated states rather than the resting state. This behavior allows these drugs to block rapidly firing or diseased tissues preferentially, sparing healthier tissues that spend more time in the resting state.
Classification of Sodium Channel Blockers
1. Antiarrhythmic Sodium Channel Blockers (Class I Agents)
In the Vaughan Williams classification of antiarrhythmic drugs, Class I agents are subdivided according to their electrophysiological effects:
- Class IA: Moderately depress upstroke (phase 0) and prolong action potential duration. Examples: Quinidine, Procainamide, Disopyramide.
- Class IB: Mildly or minimally depress phase 0; may shorten action potential duration. Examples: Lidocaine, Mexiletine.
- Class IC: Markedly depress phase 0; minimal effect on action potential duration. Examples: Flecainide, Propafenone.
These differences reflect distinct kinetics of binding and unbinding to sodium channels, influencing how effectively they act on rapidly depolarizing cardiac cells.
2. Local Anesthetics
Local anesthetics like Lidocaine, Bupivacaine, and Ropivacaine block peripheral nerve conduction by binding primarily to Naᵥ1.7 and Naᵥ1.8 channels. This prevents the generation and propagation of sensory (and sometimes motor) nerve impulses. The structure typically includes a hydrophobic aromatic ring connected to a tertiary amine via an intermediate chain. Agents differ in potency, duration, and risk of cardiotoxicity or neurotoxicity.
3. Anticonvulsants
Numerous anticonvulsants (e.g., Phenytoin, Carbamazepine, Lamotrigine) inhibit high-frequency neuronal firing by stabilizing sodium channels in their inactivated state. This effect lowers neuronal excitability and reduces the likelihood of seizure discharges, particularly in partial and tonic-clonic epilepsy.
4. Anti-Pain Medications
In addition to local anesthetics, some agents like Mexiletine (traditionally a Class IB antiarrhythmic) or certain atypical anticonvulsants are employed off-label or for neuropathic pain by moderating peripheral nociceptive sodium channels. This synergy between analgesia and antiarrhythmic/anticonvulsant strategies demonstrates the broad applications of sodium channel modulators.
Mechanism of Action and Kinetics
State-Dependent Binding and Use-Dependence
Many sodium channel blockers exhibit a preference for channels in the open or inactivated states (i.e., under conditions of frequent depolarization). This characteristic, often termed use-dependence, means that the higher the firing rate, the more effectively the drug blocks the channel.
- Use-dependence also explains the rationale behind antiarrhythmic usage in tachyarrhythmias or anticonvulsant usage in high-frequency seizure discharges.
Voltage-Dependence
Certain drugs shift the inactivation curve of the sodium channel so that a smaller fraction of channels is available at normal resting membrane potentials. This shift can stabilize membranes against depolarization, further dampening excitability.
Recovery from Block
After repolarization or non-depolarized intervals, sodium channels revert to the resting state. Drugs with slow dissociation constants remain bound longer, prolonging blockade even at normal heart rates (as seen with Class IC antiarrhythmics). In contrast, fast-dissociating drugs (e.g., lidocaine) primarily block sodium channels during high-frequency activity yet unbind when the cell returns to normal sinus pacing.
Clinical Applications of Sodium Channel Blockers
1. Cardiac Arrhythmias
Class IA
- Quinidine: Historically used for atrial fibrillation or flutter and certain ventricular arrhythmias. Also exerts alpha-adrenergic blocking effects, potentially causing hypotension.
- Procainamide: Useful for both supraventricular and ventricular tachyarrhythmias but can cause drug-induced lupus.
- Disopyramide: Has prominent negative inotropic and anticholinergic actions, sometimes used in hypertrophic obstructive cardiomyopathy.
Class IB
- Lidocaine: Preferred agent for acute ventricular tachycardia or post-MI arrhythmias. Low proarrhythmic potential with minimal effect on conduction in healthy tissue.
- Mexiletine: An oral analog of lidocaine employed for chronic ventricular arrhythmias and neuropathic pain.
Class IC
- Flecainide: Powerful suppressor of phase 0 upstroke, utilized for atrial fibrillation (including chemical cardioversion) or life-threatening ventricular arrhythmias. However, caution arises from its proarrhythmic risk in ischemic heart disease.
- Propafenone: Similar to flecainide with additional mild beta-blocking effect; used in atrial fibrillation or supraventricular tachycardias.
2. Local Anesthesia
- Lidocaine: Gold-standard local anesthetic for infiltration, nerve blocks, and epidural anesthesia. Rapid onset and intermediate duration.
- Bupivacaine: Longer-acting, more potent, but also more cardiotoxic in overdose. Used in procedures needing extended pain relief (e.g., post-op analgesia for labor and spinal anesthesia).
- Ropivacaine: Shares similarities with bupivacaine but generally less cardiotoxic. Preferred for prolonged regional blocks.
- Articaine: Popular dental anesthetic with better bone penetration.
3. Epilepsy
- Phenytoin: Effective in tonic-clonic and partial seizures. At higher levels, causes nystagmus, ataxia, gum hyperplasia, and other side effects.
- Carbamazepine: Mainstay for partial seizures, also beneficial in trigeminal neuralgia. Induces hepatic enzymes.
- Lamotrigine: Broad antiseizure coverage, used in partial, generalized tonic-clonic, and absence seizures. Minimal sedation with favorable side-effect profile.
- Valproate (multi-mechanistic): Also partially blocks sodium channels while enhancing GABA. Treats various seizure types and bipolar disorder.
4. Neuropathic Pain
- Mexiletine: Oral local anesthetic analog, sometimes used in diabetic neuropathy or nerve injury-related pain.
- Carbamazepine: Alleviates trigeminal neuralgia, reflective of its sodium channel inhibition in central pain pathways.
5. Other Uses
- Class IB antiarrhythmics have roles in advanced life support protocols, though amiodarone often replaces lidocaine in many guidelines.
- Some atypical or investigational sodium channel blockers target specific Naᵥ isoforms in migraines or demyelinating disorders (e.g., multiple sclerosis) to modulate nerve excitability.
Adverse Effects and Toxicity
1. Cardiac Effects
- Proarrhythmia: Especially with Class IC agents in ischemic hearts (the CAST trial highlighted flecainide’s risk).
- Negative Inotropy: Some can depress myocardial contractility (e.g., disopyramide, bupivacaine).
- Conduction Blocks: Excess block of His-Purkinje system or SA/AV node conduction, causing bradyarrhythmias or heart block.
2. Neurological Effects
- CNS Toxicity: Local anesthetics (e.g., lidocaine, bupivacaine) can induce seizures or confusion if plasma levels rise excessively.
- Drowsiness, Dizziness: Not uncommon with antiarrhythmics or anticonvulsants.
- Special Seizure Threshold Concerns: Some sodium channel blockers can lower the seizure threshold paradoxically if used outside intended dosing.
3. Systemic and Organ-Specific Toxicities
- Hepatic Metabolism Issues: Some cause hepatic enzyme induction (carbamazepine, phenytoin) leading to drug-drug interactions.
- Gingival Hyperplasia: A hallmark of phenytoin.
- Drug-Induced Lupus: Procainamide or hydralazine.
- Dermatologic Reactions: Stevens-Johnson Syndrome (rare but severe) with certain anticonvulsants (e.g., lamotrigine, carbamazepine).
4. Local Anesthetic Systemic Toxicity (LAST)
Occurs when local anesthetic inadvertently enters the systemic circulation (e.g., vascular puncture). Can manifest with CNS excitation (seizures) followed by CNS depression, then cardiovascular collapse. Bupivacaine is notably cardiotoxic. Intralipid infusion is a recognized rescue therapy for severe LAST.
Pharmacokinetic Considerations
Absorption and Distribution
- Oral Bioavailability: Many antiarrhythmic sodium channel blockers (e.g., mexiletine, flecainide) have moderate-to-high oral bioavailability. Lidocaine, however, undergoes extensive first-pass metabolism, so it’s given IV for arrhythmias.
- BBB Penetration: Agents like lidocaine or phenytoin readily cross the blood-brain barrier, explaining CNS side effects.
Metabolism
- Hepatic Enzymes: Cytochrome P450 isoforms (e.g., 3A4, 2D6, 2C9) handle metabolism of many sodium channel blockers (quinidine, flecainide, carbamazepine).
- Active Metabolites: Some drugs form active metabolites (e.g., procainamide → N-acetyl procainamide).
- Enzyme Induction/Inhibition: Carbamazepine induces, cimetidine can inhibit metabolism. Monitoring plasma levels helps manage narrow therapeutic indices (e.g., phenytoin).
Elimination
- Primarily renal excretion of metabolites.
- Adjusting doses for reduced renal or hepatic function is vital, particularly with antiarrhythmics or phenytoin.
Clinical Pearls for Use
- Indication-Specific Selection: For example, Class IB (lidocaine) is best for acute ventricular arrhythmias in ischemic tissue, whereas Class IC is beneficial for atrial fibrillation in structurally normal hearts.
- Slow vs. Fast On-Off: A drug that dissociates from channels slowly (Class IC) is extremely potent but can cause harmful conduction delays in diseased hearts.
- Local Anesthetic Choice: Tailor potency and duration needs. Bupivacaine is potent but watch for cardiotoxicity; ropivacaine or lidocaine might be safer.
- Anticonvulsant Dosing: Taper up, carefully monitoring seizure control and side effects; watch out for sedation or hepatic enzyme induction.
- Monitoring Drug Levels: Particularly critical for phenytoin, lidocaine (IV infusion), and certain antiarrhythmics with narrow therapeutic windows.
- Toxicity Signs: Early recognition of local anesthetic systemic toxicity (CNS excitation), conduction blocks, or proarrhythmic changes on ECG can prevent severe morbidity.
Emerging and Investigational Sodium Channel Blockers
Selective Neural Isoform Blockers
Novel drug candidates aim for selective blockade of Naᵥ1.7 or Naᵥ1.8—pain-associated isoforms—potentially providing analgesia without major sedation or cardiac side effects.
Gene Therapy Approaches
Research exists into regulating sodium channel expression or function genetically—particularly for congenital arrhythmias (e.g., Brugada syndrome, Long QT type 3) or severe neuropathic pain.
Cryo or Voltage Sensor “Trapping”
Advanced molecules or biologics designed to bind the voltage sensor domain might “trap” the channel in a nonconducting state. This approach hopes to refine use-dependence for highly pathologic conditions like epilepsy or malignant arrhythmias without affecting normal tissues substantially.
Combination Therapies
Combining sodium channel inhibition with other mechanisms (e.g., calcium channel blockade, kappa agonism for pain) might optimize efficacy while reducing required doses of each agent, thus minimizing side effects.
Special Populations
Pediatric Patients
- Dosing: Must adapt to weight, immature hepatic/renal function. For local anesthesia, total safe doses must be carefully calculated.
- Anticonvulsants: Phenytoin or lamotrigine usage in children demands close monitoring, given growth changes and dynamic metabolism.
Geriatric Patients
- Reduced Metabolic Clearance: Increases half-lives, raising toxicity risk.
- Polypharmacy: Interactions more likely with chronic conditions. Adjusting dose of Class I antiarrhythmics or antiseizure medication is crucial.
Pregnancy and Lactation
- Many sodium channel blockers, if necessary, are used with caution due to potential teratogenic or neurodevelopmental impacts. For instance, certain older anti-seizure medications (e.g., valproate, phenytoin) have known teratogenicity.
Renal/Hepatic Impairment
- Dose Adjustments: Larger accumulation risk, especially with prolonged infusion (e.g., IV lidocaine in persistent vent arrhythmias).
- Monitoring: Repeated labs and possibly drug levels are recommended.
Clinical Vignette Examples
Vignette 1: Atrial Fibrillation with Structurally Normal Heart
A 55-year-old patient experiences paroxysmal atrial fibrillation with minimal underlying heart disease. Flecainide is initiated to help maintain sinus rhythm. Over time, the patient’s ECG shows a widened QRS but no bradycardia. The medication is continued with caution, especially watching for proarrhythmic events if the patient develops ischemic heart disease in the future.
Vignette 2: Post-MI Ventricular Tachycardia
A 62-year-old with recent inferior myocardial infarction develops frequent ventricular ectopy progressing to sustained VT. An IV lidocaine infusion is chosen for acute management. A continual ECG monitoring and serum drug levels help prevent toxicity. If the patient transitions to an oral antiarrhythmic, mexiletine might be used if close follow-up is feasible.
Vignette 3: Local Anesthetic Systemic Toxicity
A 30-year-old in labor receives an epidural with bupivacaine. Minutes later, she experiences tinnitus, metallic taste, followed by seizures—signs of local anesthetic systemic toxicity (LAST). Prompt action entails stopping infusion, administering benzodiazepines for seizures, and initiating intralipid therapy. Hemodynamic support is provided to ward off potential cardiac collapse.
Vignette 4: Chronic Neuropathic Pain
A diabetic patient with peripheral neuropathy fails standard analgesics. An off-label trial of mexiletine helps reduce burning foot pain. However, due to mild GI upset and occasional dizziness, dose titration is gradual. ECG monitoring rules out conduction defects before prescribing.
Future Directions and Summary
Sodium channel blockers represent a diverse and fundamental category of pharmacotherapeutic agents with applications spanning cardiology, neurology, anesthesiology, and pain management. While conventional agents like lidocaine, phenytoin, or flecainide remain clinically entrenched, ongoing research seeks to refine molecular specificity, thus enhancing efficacy while mitigating serious adverse effects—particularly cardiotoxicity, CNS toxicity, or proarrhythmia.
Advances in molecular biology and structure-based drug design promise to unravel selective sodium channel inhibitors that target disease-specific isoforms without disrupting normal physiologic channels. Such innovations may revolutionize therapies for intractable pain, multidrug-resistant epilepsy, or refractory arrhythmias. Until then, meticulous dosing, patient-specific selection, and close monitoring underscore the safe and effective employment of sodium channel blockers.
References
- Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 13th Edition
- Katzung BG, Basic & Clinical Pharmacology, 15th Edition
- Rang HP, Dale MM, Rang & Dale’s Pharmacology, 8th Edition







[…] Sodium Channel Blockers (SCBs) […]
[…] Class I: Sodium channel blockers […]