

Pharmacokinetics, derived from the Greek words ‘pharmakon’ meaning drug, and ‘kinesis’ meaning movement, is the science that elucidates what the body does to a drug following administration. Pharmacokinetics is the study of how drugs move through the body. It includes processes like absorption, distribution, metabolism, and excretion (ADME), which determine how much of the drug is present at its action site and how quickly it leaves the body. This chapter will comprehensively explore these pharmacokinetic principles, underlining their critical importance in drug development, dosage regimen design, and therapeutic application.
“What the body does to the drug“
Absorption
Absorption is the initial stage in the pharmacokinetic pathway, where the drug transitions from the site of administration into systemic circulation. The route of administration (oral, intravenous, topical, etc.), the physicochemical characteristics of the drug (like solubility and stability), and physiological factors (like gastric pH and motility) can all affect the rate and extent of drug absorption.
The following are the different types of absorption mechanisms:

Passive diffusion
Passive diffusion is the most prevalent drug absorption mechanism. It occurs when a drug migrates from a region of high concentration to a low concentration, fueled by the concentration gradient. The concentration gradient is the variance in drug concentration between the administration site and the bloodstream. The greater the concentration gradient, the quicker the drug gets assimilated. Passive diffusion occurs via the lipid bilayer of cell membranes. The drug molecules must be lipid-soluble to permeate the cell membrane. Water-soluble molecules necessitate transporters to traverse the cell membrane. Several factors influence passive diffusion, including the drug’s molecular weight, lipid solubility, pH, and surface area of the absorption site. Bigger molecules and those with lower lipid solubility have a slower rate of absorption.
Facilitated diffusion
Facilitated diffusion is a type of passive diffusion that requires a carrier protein to carry the drug across the cell membrane. Unlike passive diffusion, facilitated diffusion is a carrier-mediated transport system. It is dependent on the concentration gradient but is limited by the number of available transporters. Facilitated diffusion is applicable for drugs that are water-soluble or too large to cross the cell membrane. The transporter proteins bind to the drug molecule and carry it across the membrane.
Active transport
Active transport is a type of transport that requires energy as the drug moves against the concentration gradient. It involves the use of carrier proteins that necessitate energy to move the drug from an area of low concentration to an area of high concentration. Active transport is useful for drugs that are required in high concentrations in specific body areas. This mechanism is commonly used for drugs that target the brain or other organs with low drug permeability.
Endocytosis and exocytosis
Endocytosis occurs when a cell membrane engulfs a drug molecule and internalizes it into the cell. This mechanism is used for large molecules or those that cannot cross the cell membrane. Once the drug is inside the cell, it can be transported to the bloodstream through various intracellular transport mechanisms. Exocytosis is the opposite process, where a cell releases a drug molecule outside of the cell. Cells commonly use this mechanism to release waste or molecules that are too large to cross the cell membrane.
Factors affecting drug absorption
Several factors can affect drug absorption, including:
Physicochemical properties of the drug: The drug’s physicochemical properties have a pivotal role in its absorption into the system. Solubility, in particular, affects how much of the drug enters the bloodstream. More soluble drugs dissolve quickly in the gastrointestinal tract and can easily pass through cell membranes. The size and shape of the drug molecule also play a role in its absorption. Large molecules can have a difficult time passing through cell membranes. The degree of ionization of a drug affects how easily it can pass through cell membranes that are only permeable to non-ionized molecules.
Route of administration: The route of administration is another crucial factor in drug absorption. The rate and extent of absorption differ based on the administration method. Oral administration typically results in slow and incomplete absorption compared to intravenous administration. The gastrointestinal tract and liver metabolize oral drugs before they enter the systemic circulation. In contrast, intravenous administration results in rapid and complete drug absorption by bypassing the gastrointestinal tract and liver.
Presence of food or other drugs in the GIT: The presence of food or other drugs in the gastrointestinal tract can also affect drug absorption. Food can slow gastric emptying, alter the pH of the gastrointestinal tract, and reduce the absorption of some drugs. Some drugs can also interact with others, leading to changes in absorption rates and extents.
Blood flow to the site of administration: The blood flow to the site of administration also affects drug absorption. Areas with high blood flow, such as the lungs and liver, absorb drugs more rapidly than areas with low blood flow, like subcutaneous tissue.
The surface area available for absorption: The surface area available for drug absorption is another critical factor. Larger surface areas, like skin and mucous membranes, absorb drugs more quickly than smaller surface areas.
pH of the drug: The pH of the drug can also affect its absorption rate. Weak acids or bases absorb differently based on the surrounding environment’s pH. For instance, weak acids absorb more easily in acidic environments, while weak bases absorb more easily in alkaline environments. A thorough understanding of a drug’s pH and surrounding environment can enhance its absorption and efficacy.
Click here for the detailed Factors Affecting Drug Absorption page
Oral administration, the most commonly used route, involves complex absorption processes. The drug must survive the acidic stomach environment, permeate through the intestinal epithelium, and endure first-pass metabolism in the liver before reaching systemic circulation. These steps can significantly affect the bioavailability of the drug, i.e., the proportion of the administered drug that ultimately reaches the systemic circulation.
Bioavailability
Bioavailability, an integral component of pharmacokinetics, represents the fraction of an administered drug that reaches the systemic circulation unchanged. It profoundly influences a drug’s efficacy and safety. This chapter provides an in-depth exploration of bioavailability and the factors influencing it, further augmented by relevant examples.
Understanding Bioavailability
Bioavailability is a critical determinant in drug design and administration. It’s essential in establishing the dosage, route of administration, and frequency of use for a given drug. After oral administration, a drug must overcome several barriers—like degradation in the stomach, poor permeability across the intestinal wall, and first-pass metabolism in the liver—before it reaches systemic circulation. The bioavailability of a drug is expressed as a percentage of the administered dose. For instance, intravenous administration bypasses these barriers and thus has a bioavailability of 100%. Conversely, orally administered drugs often exhibit lower bioavailability. For example, the antiviral drug ribavirin has an oral bioavailability of approximately 64%.
Factors Affecting Bioavailability
Several factors, both intrinsic and extrinsic, affect a drug’s bioavailability.
Physicochemical Properties of the Drug
The physicochemical properties of a drug, such as solubility and stability, profoundly influence its bioavailability. Drugs must be soluble in bodily fluids to be absorbed. Poorly soluble drugs, like spironolactone, may have low bioavailability. Similarly, drugs that are unstable in the acidic environment of the stomach, such as penicillin G, can be degraded before absorption, reducing their bioavailability.
Dosage Form and Drug Formulation
The drug’s dosage form and formulation significantly impact its bioavailability. The size of particles in a drug formulation, their crystalline form, and the presence of excipients can influence the rate and extent of drug absorption. For instance, the antifungal drug griseofulvin has poor bioavailability due to its large crystalline structure. However, its bioavailability significantly improves when formulated as a microsize or nanosize suspension.
First-Pass Metabolism
First-pass metabolism is a phenomenon where a drug is extensively metabolized in the liver before it reaches systemic circulation. Drugs with high first-pass metabolism, such as propranolol, have reduced bioavailability when administered orally.
Physiological and Pathological Factors
Individual physiological factors like age, sex, and genetic polymorphisms can affect bioavailability. Pathological conditions, such as gastrointestinal disorders, liver diseases, or heart failure, can also influence a drug’s bioavailability. For example, liver diseases may impair first-pass metabolism, increasing the bioavailability of certain drugs.
Distribution
Following absorption, the drug disperses throughout the body’s fluids and tissues—a process known as distribution. The extent and rate of drug distribution depend on factors such as blood flow to the organs, the drug’s lipid solubility, protein binding characteristics, and the permeability of cell membranes. Some drugs bind extensively to plasma proteins, particularly albumin. This binding affects the drug’s distribution as only the free (unbound) drug can cross cell membranes to reach its site of action or undergo metabolism and excretion. Therefore, understanding a drug’s protein binding property is crucial for predicting its pharmacokinetic behaviour.
The following factors can affect drug distribution:
Plasma protein binding
Drug pharmacokinetics heavily relies on plasma protein binding. Once a drug enters the bloodstream, it can either remain unbound or bind to plasma proteins. The drug’s efficacy and toxicity are substantially affected by the amount of drug that binds to proteins versus the unbound quantity. A drug’s distribution throughout the body is significantly impacted by protein binding. If a drug is bound to proteins, it cannot penetrate cell membranes, resulting in limited distribution to extracellular fluid compartments. The small volume distribution is typical for drugs with high protein binding, which indicates confinement to the bloodstream and extracellular fluid. Nevertheless, drug interactions affect plasma protein binding. Drugs that compete for the same binding sites on proteins can displace one another, leading to changes in free drug concentrations and subsequent pharmacological effects. Similarly, the concentration of plasma proteins can affect drug binding, altering drug efficacy and toxicity. Displacement takes place when two drugs compete for the same binding site on a protein. The drug that has a higher affinity for the binding site displaces the other drug, increasing the free drug concentration and potentially causing changes in pharmacological effects. Endogenous compounds such as hormones and neurotransmitters that bind to plasma proteins can also displace drugs. Drug displacement interactions can have significant clinical consequences. If another drug displaces a highly protein-bound drug, its free concentration will increase, which may lead to potential toxicity or adverse effects. Thus, drug interactions that involve protein binding require close monitoring, particularly in patients taking multiple drugs.
Volume of distribution
The volume of distribution (Vd) is a pharmacokinetic parameter representing the apparent volume of drug distribution in the body. It is a theoretical value that determines how much of the administered drug has dispersed into body tissues relative to the concentration of the drug in the bloodstream.
The apparent Vd can be calculated using the equation:
Vd = amount of drug in the body/plasma drug concentration
This equation assumes that the drug is uniformly distributed throughout the body and that the plasma concentration accurately represents the total drug concentration in the body. The Vd can provide insights into a drug’s pharmacokinetic properties, including its distribution, metabolism, and excretion. Drugs with high Vd values are typically distributed more widely throughout the body, whereas drugs with low Vd values are more confined to the bloodstream and extracellular fluid. F
or example, a drug with a Vd of 10 L would indicate that the drug is distributed throughout a larger volume than the actual volume of the body (approximately 5-6 L for an adult).
Conversely, a drug with a Vd of 0.1 L would indicate that the drug is more confined to the bloodstream and extracellular fluid.
It is worth noting that the Vd can vary based on several factors, such as the drug’s physicochemical properties, plasma protein binding, tissue permeability, and regional blood flow. Therefore, it is essential to interpret the Vd in the context of other pharmacokinetic parameters to fully understand a drug’s disposition and optimize its dosing regimen.
Tissue and organ barriers
Some organs, such as the brain and placenta, have specialized barriers that prevent drugs from entering.
Metabolism
Drug metabolism predominantly occurs in the liver, which serves as the primary site for the biotransformation of drugs and other xenobiotics. The liver contains a variety of enzymes that modify the chemical structure of drugs, typically rendering them more water-soluble and easier to excrete from the body. The most important family of enzymes involved in drug metabolism is the cytochrome P450 (CYP) enzymes. However, drug metabolism can also occur in other tissues and organs, including:
- Intestines: Some drugs are metabolized in the intestines before they even reach systemic circulation, a phenomenon known as the first-pass effect or first-pass metabolism.
- Kidneys: The kidneys play a role in both the metabolism and excretion of drugs, especially for compounds that are renally cleared.
- Lungs: The lungs can metabolize some inhaled drugs and other substances.
- Skin: Topically applied drugs can undergo metabolism in the skin.
- Brain: Some drugs can be metabolized in the brain, affecting their psychoactive properties.
- Plasma: Some drugs are metabolized in the blood plasma by plasma esterases.
Drug metabolism is a pivotal aspect of pharmacology and toxicology, dictating a given drug’s pharmacokinetics and toxicological impact within a biological system. It primarily involves chemical reactions that transform drugs into more polar, water-soluble metabolites, facilitating their elimination from the body. Drug metabolism, also known as biotransformation, is a process that converts drugs into more polar metabolites for easier excretion. Metabolism involves various enzymes, primarily occurring in the liver, chiefly the cytochrome P450 (CYP450) enzyme family. The rate of metabolism can significantly impact a drug’s pharmacological activity. Some drugs, known as prodrugs, are inactive or less active until metabolized into their active forms. Conversely, metabolism can render some drugs inactive or even produce toxic metabolites. Drug-drug interactions are a significant concern during metabolism. When the same enzyme metabolizes two drugs, they can compete for the enzyme, leading to altered drug levels in the body, which can result in therapeutic failure or toxicity.
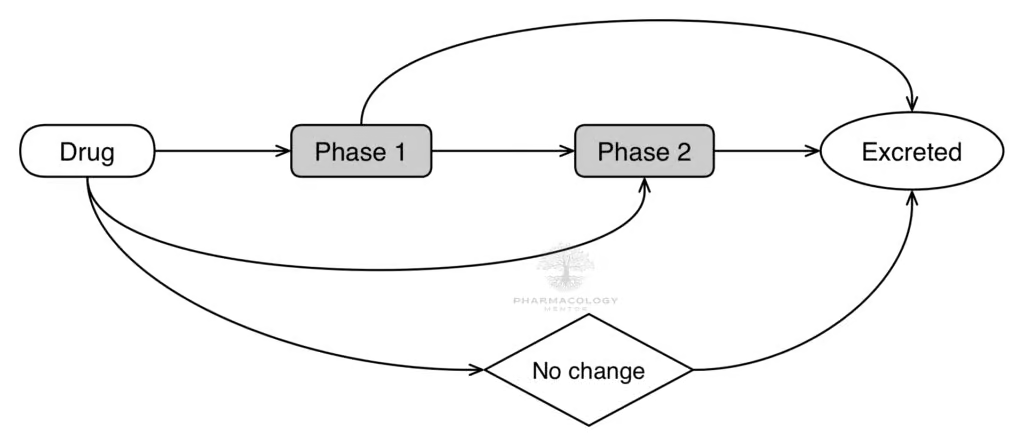
There are two phases of drug metabolism:
Phase I reactions: Oxidation, reduction, and hydrolysis
Phase I reactions, also known as functionalization reactions, are the initial step in the biotransformation of drugs. These reactions primarily involve oxidation, reduction, and hydrolysis, with the aim to increase the drug’s hydrophilicity, thereby facilitating further metabolism in Phase II reactions or direct excretion. It’s worth noting that Phase I reactions can sometimes render drugs more pharmacologically active or toxic than their original forms. Cytochrome P450 (CYP450) enzymes, a diverse family of heme-thiolate monooxygenases predominantly located in the liver, play a crucial role in these reactions. For instance, CYP3A4, one of the most abundantly expressed CYP450 enzymes, metabolizes approximately 50% of clinically used drugs, including statins and calcium channel blockers.
Phase I reactions can occur through three mechanisms:
Oxidation Reactions
Oxidation reactions represent the most common type of Phase I reactions. These reactions introduce an oxygen atom into the drug molecule, usually forming a new hydroxyl group. Oxidation reactions are primarily catalyzed by the cytochrome P450 (CYP450) enzyme family, which contains over 50 different isoforms. CYP3A4, one of the most abundantly expressed CYP450 enzymes, is known to metabolize a wide range of drugs. For instance, it metabolizes atorvastatin, a commonly prescribed statin for the management of hypercholesterolemia, through hydroxylation. Another example includes the metabolism of the benzodiazepine alprazolam, where CYP3A4 catalyzes the hydroxylation of the drug, forming alpha-hydroxyalprazolam, which is still pharmacologically active but less potent than the parent drug.
Reduction Reactions
Reduction reactions involve the addition of hydrogen atoms to the drug molecule or the removal of oxygen atoms. These reactions typically occur on molecules with nitro, azo, or carbonyl groups and are usually catalyzed by enzymes such as cytochrome P450 reductases or nitroreductases. An illustrative example is the biotransformation of the nitrate drug isosorbide dinitrate, used to treat angina pectoris. It is reduced to its mononitrate metabolites, which are active and responsible for the drug’s anti-anginal effects.
Hydrolysis Reactions
Hydrolysis reactions involve breaking a chemical bond in the drug molecule by adding a water molecule. These reactions typically occur on ester or amide bonds and are catalyzed by esterases or amidases. An example of hydrolysis can be found in the metabolism of the drug aspirin (acetylsalicylic acid). Aspirin is hydrolyzed by esterases in the plasma and liver to produce salicylic acid, the active metabolite responsible for the drug’s anti-inflammatory and analgesic effects.
Phase II reactions: Conjugation
Subsequent to Phase I, Phase II reactions involve the conjugation of the drug or Phase I metabolite with endogenous substrates such as glucuronic acid, sulfate, or glutathione. This substantially increases the metabolite’s polarity, enhancing its renal excretion. For example, acetaminophen, a common over-the-counter analgesic and antipyretic, undergoes Phase II glucuronidation and sulfation predominantly, resulting in harmless, water-soluble metabolites excreted via urine.
The most common conjugation reactions include:
Glucuronidation
Glucuronidation is the most common Phase II reaction, which involves the addition of a glucuronic acid moiety to the drug or its metabolite. This reaction is catalyzed by the family of enzymes known as uridine diphosphate-glucuronosyltransferases (UGTs). A classic example of glucuronidation is seen in the metabolism of paracetamol (acetaminophen). After a small fraction of paracetamol undergoes Phase I metabolism, the majority is converted into a harmless glucuronide conjugate which is then excreted in the urine.
Sulfation
Sulfation involves the addition of a sulfate group to the drug or metabolite, a reaction catalyzed by sulfotransferase (SULT) enzymes. Sulfation also plays a role in the metabolism of paracetamol. Alongside glucuronidation, sulfation forms a harmless sulfate conjugate which, like the glucuronide conjugate, is excreted in the urine.
Acetylation
Acetylation involves the transfer of an acetyl group from acetyl-CoA to the drug or metabolite. N-acetyltransferase (NAT) enzymes catalyze the reaction. A well-known example of acetylation is the biotransformation of isoniazid, a first-line antitubercular drug. NAT2 acetylates Isoniazid into acetylisoniazid, which is less active and further metabolized before being excreted.
Methylation
Methylation involves the transfer of a methyl group from S-adenosyl methionine (SAM) to the drug or metabolite. Various methyltransferases catalyze this reaction. An example of methylation is seen in the metabolism of the neurotransmitter dopamine, where it is methylated by catechol-O-methyl transferase (COMT) to form 3-methoxytyramine, which is less biologically active.
Glutathione Conjugation
Glutathione conjugation involves the addition of a glutathione molecule to the drug or metabolite, a reaction catalyzed by glutathione S-transferase (GST) enzymes. A key example of glutathione conjugation is seen again with paracetamol. CYP450 enzymes convert a small fraction of paracetamol into a toxic metabolite (NAPQI) during Phase I metabolism. This toxic metabolite is then rapidly conjugated with glutathione to form a non-toxic compound that can be safely excreted.
Drug-Metabolizing Enzymes
The efficiency and outcome of drug metabolism are significantly influenced by the functional profile of the host’s drug-metabolizing enzymes. Genetic polymorphisms can cause interindividual variations in enzyme activity, leading to variable drug responses among different people.
Genetic Polymorphisms and Drug Metabolism
To illustrate, polymorphisms in CYP2D6—a key enzyme metabolizing approximately 25% of clinical drugs including antidepressants, antipsychotics, and beta-blockers—lead to a wide range of metabolic phenotypes, from poor to ultrarapid metabolizers. This variability may necessitate dosage adjustments or alternative therapy selections to avoid adverse drug reactions or therapeutic failure.
Drug-Drug Interactions and Metabolism
Drug-drug interactions (DDIs) significantly impact drug metabolism, potentially altering a drug’s therapeutic effect or toxicity profile. These interactions often involve the competitive inhibition or induction of drug-metabolizing enzymes. For instance, ketoconazole, an antifungal agent, inhibits CYP3A4, potentially increasing the plasma concentrations of concurrently administered CYP3A4 substrates like simvastatin, leading to potential statin-associated myopathy.
Drug-Drug Interactions and Metabolism
Drug-drug interactions (DDIs) significantly impact drug metabolism, potentially altering a drug’s therapeutic effect or toxicity profile. These interactions often involve the competitive inhibition or induction of drug-metabolizing enzymes. For instance, ketoconazole, an antifungal agent, inhibits CYP3A4, potentially increasing the plasma concentrations of concurrently administered CYP3A4 substrates like simvastatin, leading to potential statin-associated myopathy.
Enzyme induction and inhibition
Some drugs can induce or inhibit the activity of drug-metabolizing enzymes, which can affect the metabolism and elimination of other drugs. Enzyme induction refers to the increase in the activity of drug-metabolizing enzymes, while enzyme inhibition refers to the decrease in the activity of these enzymes. Enzyme induction and inhibition can lead to changes in drug efficacy and toxicity.
Genetic polymorphisms
Genetic differences in drug-metabolizing enzymes can affect how quickly or slowly a drug is metabolized, leading to differences in drug efficacy and toxicity. Genetic polymorphisms can affect the expression or activity of drug-metabolizing enzymes, leading to inter-individual variability in drug metabolism. The most well-known genetic polymorphisms are those affecting the cytochrome P450 enzymes.
Excretion
Excretion is the final phase in the pharmacokinetic process, where the drug and its metabolites are eliminated from the body. This elimination primarily occurs via the kidneys, resulting in urinary excretion, but it can also occur through other routes such as the lungs, bile, sweat, and breast milk. Understanding the excretion rate is vital for determining the dosing interval of drugs. If the excretion rate is slow, the drug may accumulate in the body, leading to potential toxicity. In contrast, a rapid excretion rate may require more frequent dosing to maintain therapeutic drug levels. Drugs can be excreted through various routes, including urine, feces, sweat, saliva, and breath.
Renal excretion
Renal excretion is the most common route of drug elimination. It involves the filtration of drugs by the kidneys and their subsequent excretion in the urine. The kidneys filter drugs based on their size, charge, and lipophilicity. Water-soluble drugs and drugs that are metabolized to water-soluble metabolites can be excreted in the urine. Renal excretion involves glomerular filtration, passive tubular reabsorption, and active tubular secretion. Many factors can affect these processes, including urinary pH, renal blood flow, and the drug’s physicochemical properties.
Mechanism of Renal Excretion
The process of renal excretion involves three steps: filtration, reabsorption, and secretion. The drug molecules are filtered from the blood through the glomeruli and into the renal tubules. Some drugs can be reabsorbed back into the blood, reducing their elimination. However, some drugs can be actively secreted from the blood into the tubules, increasing their elimination.
Factors Affecting Renal Excretion
Several factors can affect renal excretion, including the drug’s physicochemical properties, pH of the urine, and renal function. Highly protein-bound drugs are less likely to be filtered by the kidneys and, therefore, have a longer half-life. Additionally, the pH of the urine can affect the ionization of drugs, altering their excretion. Finally, renal function plays a crucial role in drug excretion because impaired kidney function can lead to a longer elimination half-life.
Biliary excretion
Biliary excretion is another route of drug elimination. Some drugs can be eliminated through the bile into the feces, which can then be excreted from the body. This route is especially important for highly lipophilic drugs that cannot be excreted through the kidneys.
Other routes of excretion
Depending on their physicochemical properties, some drugs can be eliminated through sweat, saliva, or breath. However, these routes of excretion are less common than renal or biliary excretion.
Sweat
Some drugs can be eliminated through sweat, primarily those that are highly lipophilic and can penetrate the skin. However, the amount of drug eliminated through sweat is typically small compared to renal or biliary excretion.
Saliva
Some drugs can be eliminated through saliva, primarily those that are apolar and can diffuse across the salivary glands. However, this route of excretion is not significant compared to renal or biliary excretion.
Breath
Some drugs can be eliminated through breath, primarily those that are volatile and can be exhaled through the lungs. This route of excretion is typically only relevant for drugs that are inhaled, such as anesthetics.
Conclusion
Pharmacokinetics is a vital aspect of drug development and clinical pharmacology, as it helps us understand how drugs move through the body and how they are eliminated. By understanding the different mechanisms of drug absorption, distribution, metabolism, and elimination, we can optimize drug therapy and reduce the risk of adverse effects.
Disclaimer: This article is for informational purposes only and should not be taken as medical advice. Always consult with a healthcare professional before making any decisions related to medication or treatment.
FAQs
What is the difference between pharmacokinetics and pharmacodynamics?
Pharmacokinetics is the study of what the body does to drugs, while pharmacodynamics is the study of what drugs do to the body.
How is drug absorption affected by food?
Food can affect drug absorption by altering the pH of the gastrointestinal tract, slowing down or speeding up gastric emptying, or interacting with the drug molecules.
What is the first-pass effect?
The first-pass effect is the metabolism of a drug by the liver before it enters the systemic circulation, which can affect the bioavailability of the drug.
How can genetic polymorphisms affect drug metabolism?
Genetic polymorphisms can lead to variations in the activity of drug-metabolizing enzymes, affecting the rate and extent of drug metabolism and leading to differences in drug efficacy and toxicity.
What are the factors that affect drug distribution?
Drug distribution can be affected by plasma protein binding, volume of distribution, and tissue and organ barriers.








[…] Pharmacokinetics […]
[…] Pharmacokinetics – What the body does to Drug? […]