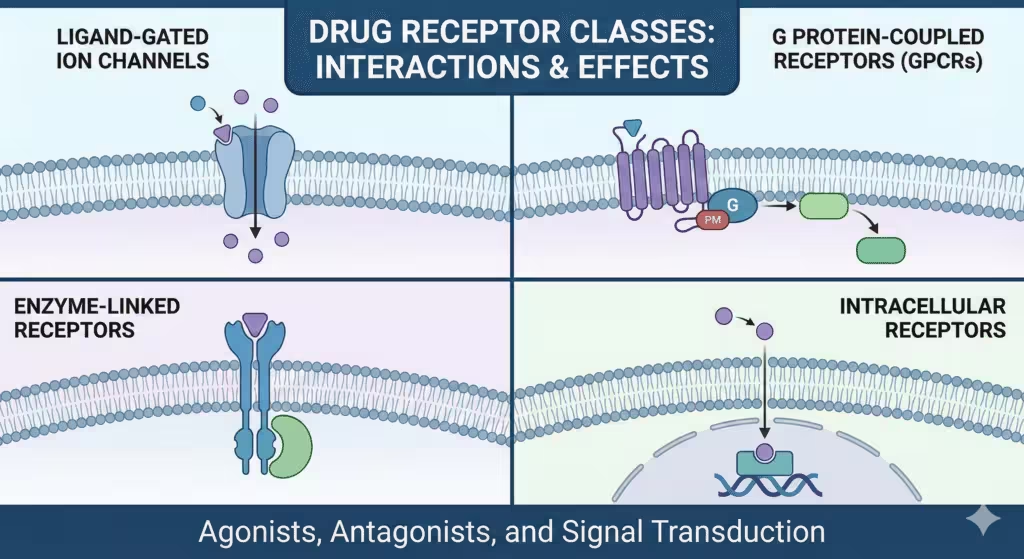
There are four classes of drug receptors, G protein-coupled receptors, ligand-gated ion channels, enzyme-linked receptors (receptor tyrosine kinases and cytokine receptors), and intracellular nuclear receptors, each of which transmits its signals through different, but well-defined pathways, from ligand binding to cellular action.
ⓘ Quick overview
Most receptor pharmacology is organized into four classes: ligand-gated ion channels, GPCRs, tyrosine kinase-coupled/enzyme-linked receptors and intracellular nuclear receptors, which is the organization that is widely used in clinical pharmacology and anesthesiology education.
In practice, these classes are distinguished by location (membrane vs intracellular), coupling (ions, G proteins, kinases, transcription), and kinetics (milliseconds to hours), which have a direct correlation to therapeutic onset and duration of action.
⚡ Ligand-gated ion channels
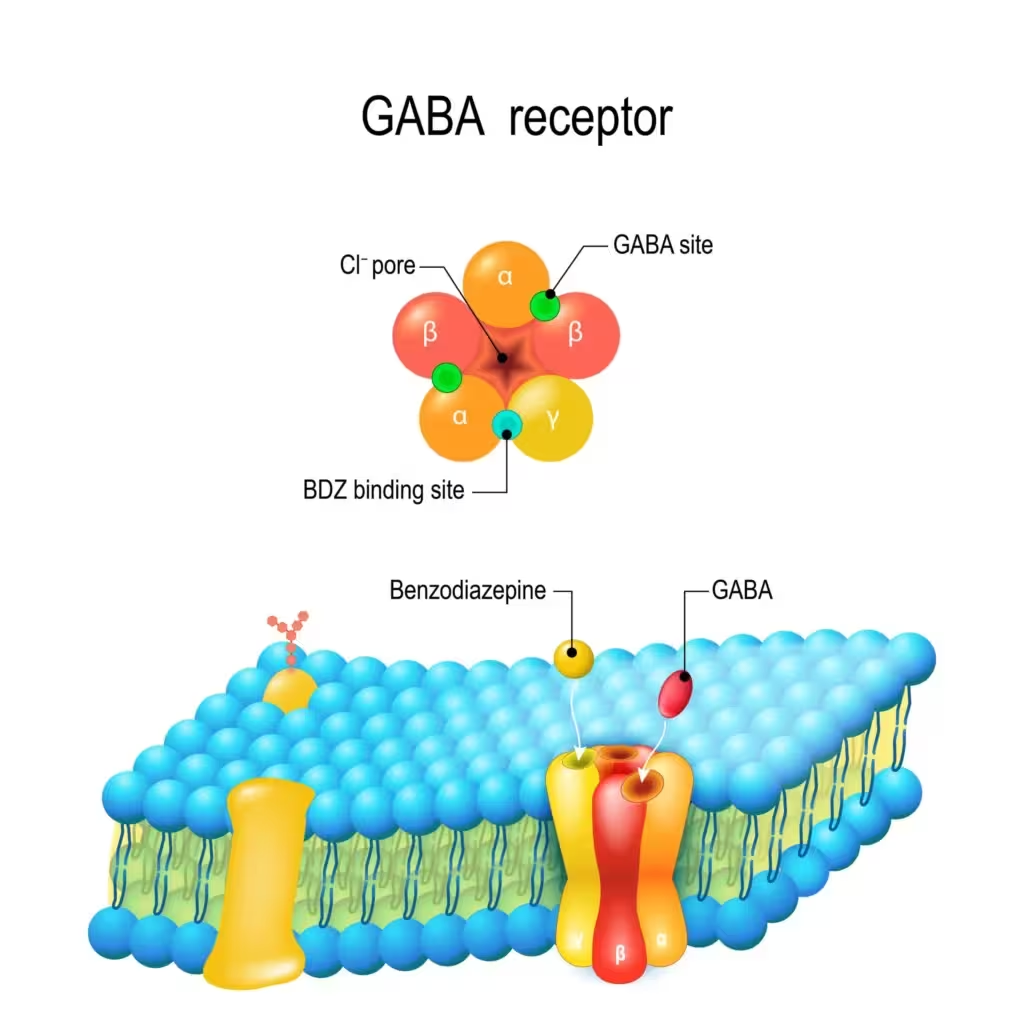
Ligand-gated ion channels (LGICs, ionotropic receptors) are transmembrane proteins that, when bound by an orthosteric ligand, open a selective pore to ions like Na+, K+, Ca2+ or Cl- and thus convert during milliseconds, the chemical signals of neurotransmitters to the swift electrical responses.
Typical LGICs are the nicotinic acetylcholine receptor and the GABA_A receptor. LGICs mediate fast synaptic transmission at distal synapses and central junctions; gating and allostery allow phasic or tonic signaling, depending on the receptor localization and local transmitter concentration.
Some channel proteins support ion selectivity (depolarization – excitatory or hyperpolarization – inhibitory) and many LGICs also comprise allosteric receptors for modulators or blockers, providing an understanding of the mode of action of drugs such as benzodiazepine potentiation via the GABA_A receptor and the NMDA channel block by ketamine.
⚙️ Mechanistic steps
Orthosteric interaction of the ligand at the extracellular domain causes a conformational change and propagates to the transmembrane domain resulting in pore gating, a phenomenon called gating isomerization which structurally separates binding of the ligand with the channel opening.
Allosteric ligands and endogenous modulators can exist across the gating equilibria, modulate open probability, and/or desensitization, thereby explaining pharmacological profiles and therapeutic windows in different therapeutic agents of the channel target sites.
🩺 Clinical implications
Because action of LGICs is within milliseconds, drugs acting at LGICs have rapid onset (e.g. neuromuscular blockers at nicotinic receptors; anaesthetics at GABA_A/NMDA), and antagonists act either as competititors at the orthosteric site or as noncompetitors blocking the channel pores (portioned reversal strategies, safety profiling).
Response modifiers such as a decrease in responsiveness to prolonged treatment due to the desensitization of receptors and variation in subunit composition across various tissues affect pharmacologic sensitivity and adverse effect profiles.
🛠️ G protein coupled receptors (GPCRs)

GPCRs are seven transmembrane receptors and also guanine nucleotide exchange factors for heterotrimeric G proteins, and GPCR stimulation leads to stabilization of an active conformation, which then catalyzes the exchange of GDP and GTP on Ga, resulting in Ga-Gbg dissociation and activation of downstream effectors and second messengers.
The four canonical Gα classes—Gs, Gi/o, Gq/11, and G12/13—link GPCRs to cAMP/PKA, inhibition of adenylyl cyclase, PLCβ→IP3/DAG/Ca²⁺/PKC, and Rho GTPase signaling, respectively, producing diverse cellular outcomes from metabolism to contractility.
GPCR signaling has bimodal aspects: G protein-domain dependent signaling is performed in parallel with β-arrestin dependent signaling that occur to mediate desensitization, endocytosis and unique interactions to kinase signaling (e.g., ERK scaffolding) such that “biased” signaling profiles are thus possible on a ligand by ligand basis.
🛡️ Desensitization and β‑arrestins
Upon repeated or sustained agonism, GPCR kinases (GRKs) phosphorylate active receptors, promoting β‑arrestin binding that sterically prevents further G protein coupling and scaffolds enzymes (e.g., PDE4, DGKs) to dampen second messengers, a core mechanism of acute desensitization.
β‑arrestins also assemble signaling complexes (e.g., ERK, JNK, Src) at the plasma membrane or endosomes, producing sustained, spatially restricted signals distinct from transient nuclear ERK waves driven by G proteins, which underlies functional selectivity of GPCR responses.
⚖️ Biased agonism
Ligands can selectively stabilize receptor conformations that preference to the G protein vs b-arrestin pathways (or vice versa), creating therapeutic opportunity to increase desired clinical effects while mitigating adverse events as exemplified by novel GPCR-targeting analgesics and cardiometabolic agents currently in development.
GRK isoform-selective phosphorylation of C-tails encodes b-arrestin conformations and functions and the selectivity of GRK isoform-mediated bias and trafficking depends on cell type, receptor C-tail sequence, and ligand chemistry.
🗂️ Enzyme linked receptors: RTKs & cytokine receptors
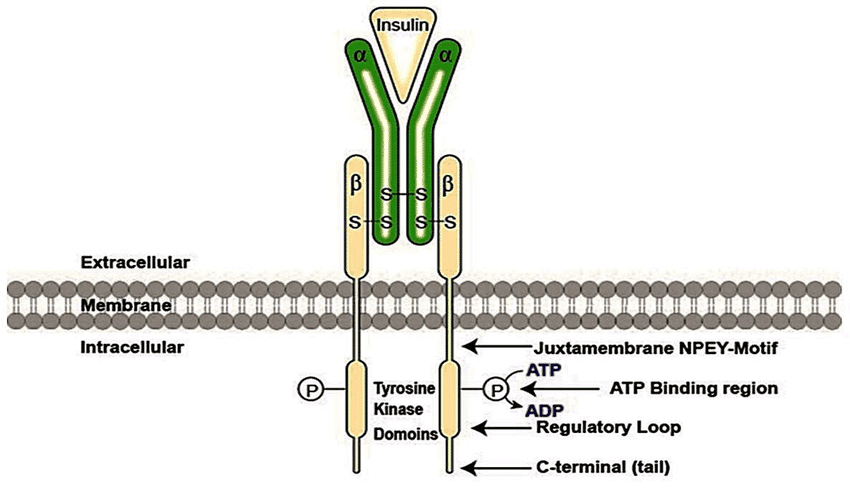
Enzyme‑linked receptors include receptor tyrosine kinases (RTKs) with intrinsic kinase domains and cytokine receptors that signal via non‑receptor tyrosine kinases such as JAKs, both converting extracellular growth factor or cytokine binding into phosphorylation cascades and transcriptional reprogramming.
RTKs (e.g., EGFR, VEGFR, PDGFR) are activated when ligand binding stabilizes receptor dimerization or oligomerization, enabling trans‑autophosphorylation on specific tyrosines that both activate the kinase and create SH2/PTB docking sites for adaptor proteins and enzymes.
The phosphorylated tail recruits effectors to Ras–MAPK, PI3K–Akt, and PLC‑γ pathways, coordinating proliferation, differentiation, survival, angiogenesis, and motility, while the precise tyrosine motif context confers pathway specificity.
📝 RTK mechanism step-by-step
Ligand binding exposes or stabilizes a dimerization interface, RTK protomers pair, and their kinase domains trans‑autophosphorylate activation loops and C‑terminal tails, switching on catalytic activity and building high‑affinity docking sites for SH2/PTB domain proteins such as Grb2, Shc, PI3K, and PLC‑γ.
Adaptor engagement triggers cascades: Grb2–SOS activates Ras→Raf→MEK→ERK (MAPK), PI3K generates PIP3 to recruit PDK1/Akt (survival/growth), and PLC‑γ hydrolyzes PIP2 to IP3/DAG to mobilize Ca²⁺ and activate PKC, integrating signals by context to yield distinct cellular outcomes.
🦠 Cytokine receptors and STAT
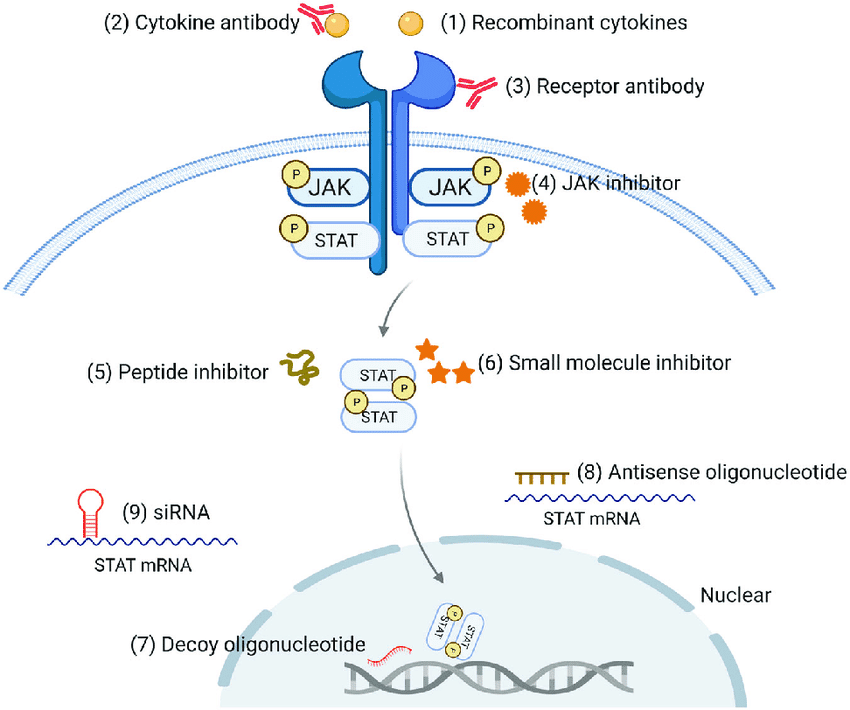
Cytokine receptors have no kinase activity, but constitutively bind to Janus kinases (JAKs); cytokine binding causes receptor juxtaposition that leads to the trans-activation of JAKs, which, in turn, phosphorylate receptor tails, leading to the recruitment of the transcription factors of the signal-dependent transcription factor family (STATs), as well as their phosphorylation, dimerization, and nuclear translocation.
JAK-STAT signaling is parsimonious – receptor, kinase and transcription factor – but powerful, directing the development, proliferation, and effector functions of immune cells with negative feedback controls to ensure the termination of a signal at the right time.
⚛️ Intracellular (nuclear) receptors
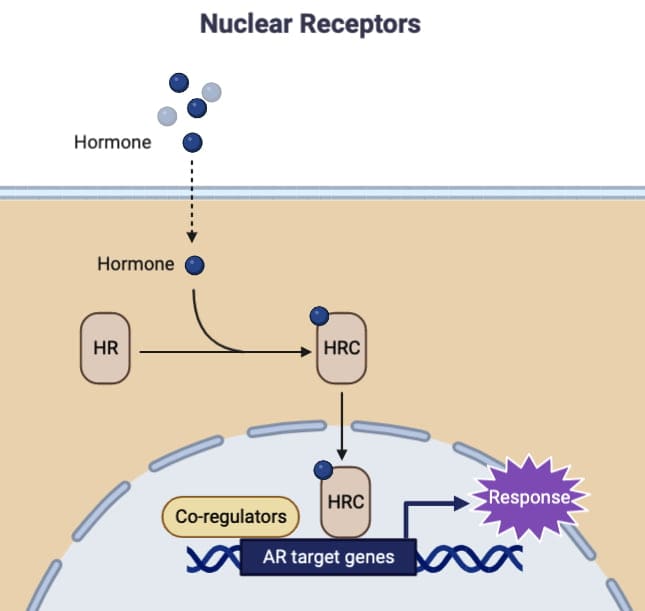
Slower onsets, and durable genomic effects, may be explained by slower effects of nuclear receptors, which are nuclear transcription factors which bind a variety of lipid soluble hormones (e.g. glucocorticoids, estrogens, thyroid hormone, retinoids), dimerize and regulate gene transcription via chromatin associated hormone response elements.
Coactivators (e.g. p160 family, CBP/p300) and corepressors are key coregulators which are recruited or released by nuclear receptors depending upon the type of ligand, and coactivators often contain histone acetyltransferase activity which serves to loosen chromatin structure to promote the initiation of transcription.
While binding to agonist usually leads to coactivator recruitment through LXXLL motifs and to chromatin remodeling, binding to antagonists or selective modulators can result in corepressor complex stabilization to downregulate target genes, and consequently lead to tissue selective pharmacology.
🛤️ Mechanistic flow
Unliganded steroid receptors can be located in the cytosol complexed with chaperones or in the nucleus. Ligand binding elicits conformational changes, dissociation from chaperone, nuclear translocation (if required), dimerization, DNA binding to response elements and coactivator recruitment for transcriptional activation.
The balance of AF-1 and AF-2 transactivation domains, receptor isoforms as well as the availability of coregulators determine the effects on genes and the basis for selective receptor modulators in oncology, endocrinology, and women’s health.
⏰ Kinetics and timescales
LGIC signaling occurs at the speed of milliseconds and is ideal for synaptic transmission that matches the clinical rapidity that agents which modulate these channels such as anesthetics and neuromuscular blockers.
GPCR signaling takes place over seconds to minutes, by means of the second messengers and protein kinase cascades, followed by desensitization and internalization to regulate length and ablation on repeated dosing.
RTK and JAK-STAT signals usually develop over the course of minutes to hours and with transcriptional outputs whereas nuclear receptor-mediated genomic actions often require hours to days to convert transcriptional programs into phenotypic alteration consistent with therapeutic latenties of endocrine drugs.
🎚️ Regulation and adaption
All receptor classes have some form of regulation like desensitization, downregulation, or supersensitivity, e.g., for GPCRs, in the form of binding of GRK to receptors and binding of a-arrestin acutely uncouples receptors from G proteins and alters the trafficking, thus influencing tolerance and biased outcomes.
RTKs are negatively regulated by phosphatases, ubiquitin ligases and endocytosis in amplitude and duration modulation; and nuclear receptor outputs are regulated by levels of coregulators, and both post-translational modifications and the chromatin environment, explaining tissue-specific pharmacology.
🔬 Clinical examples by class
LGICs: neuromuscular junction nicotinic acetylcholine receptor; competitive antagonists (e.g., NMBAs) and reversing (indirectly or with encapsulating agents) and GAB(A)A potentiation (sedative-hypnotics); potassium channel receptor (GABA) – nACh channel – postsynaptic nicotinic Type 1 muscle receptor – agonist: acetylcholine; antagonist: smoking cessation and nicotinic acetylcholine receptor agonist, donepezil 23 mg/day.
GPCRs: opioid receptors (μ, κ, δ) link to Gi/o and suppress adenylyl cyclase and regulate ion channels; arrestin-mediated signaling found in adverse effect profiles and motivating G-biased analgesic development.
RTKs: Cancer and angiogenesis depend on RTK pathways; pathway map through RA/MAPK/PI3K/Akt elucidates mechanisms of resistance and combination strategies.
Cytokine Receptors: Components of the JAK/STAT signaling pathway, JAK inhibitors decrease hyperactivated JAK-STAT signaling in inflammation and hematologic malignancies by blocking the kinase activation and transcription dependent STAT functions.
Nuclear receptors: glucocorticoids enlist coactivators to GREs to activate anti-inflammatory genes and repress the pro-inflammatory pathways; coregulator dynamics and chromatin remodeler dynamics play a major role in dose response and side effects.
🗂️ Comparison table
| Receptor class | Location | Primary coupling | Onset | Hallmark mechanisms | Example ligands |
| Ligand‑gated ion channel | Plasma membrane | Ion flux (Na⁺, K⁺, Ca²⁺, Cl⁻) | Milliseconds | Orthosteric gating, allosteric modulation, desensitization | ACh (nicotinic), GABA, glutamate |
| GPCR | Plasma membrane | Gα (Gs, Gi/o, Gq/11, G12/13) and β‑arrestin | Seconds–minutes | Second messengers (cAMP, IP3/DAG), GRK/β‑arrestin desensitization, biased signaling | Opioids, catecholamines, chemokines |
| Enzyme‑linked (RTK) | Plasma membrane | Intrinsic tyrosine kinase | Minutes–hours | Dimerization, trans‑autophosphorylation, SH2/PTB docking, MAPK/PI3K/PLC‑γ | EGF, PDGF, VEGF |
| Enzyme‑linked (cytokine) | Plasma membrane | JAK–STAT | Minutes–hours | JAK activation, STAT phosphorylation/dimerization, transcription | Interleukins, interferons |
| Nuclear receptor | Cytosol/nucleus | Transcriptional regulation | Hours–days | Ligand‑dependent coactivator recruitment, chromatin remodeling | Steroids, thyroid hormone, retinoids |
💡 Advanced nuances
Since many LGICs occur as heteropentamers or tetramers with subunit-specific pharmacology (“subunit-specific drugs”), channel-modulating drugs do not substantially cross-subunits but instead have subunit expression patterns that determine their clinical sensitivity, efficacy and side-effect risks.
GPCR cross‑talk can transactivate RTKs via β‑arrestin and Src, while Wnt/Frizzled GPCRs illustrate arrestin‑scaffolded canonical and noncanonical signaling, expanding GPCR influence beyond classical second messengers.
RTK docking specificity arises due to the linear sequence at the vicinity of phosphotyrosines that are recognized by SH2/PTB domains that encodes pathway selection at the receptor tail and determines different programs in biology.
JAK–STAT’s minimalistic architecture allows rapid, direct gene regulation with pathway attenuation by SOCS proteins and phosphatases guarding against hyperinflammation and malignant transformation.
Nuclear receptor tissue selectivity derives from differential coregulator expression and chromatin landscapes, enabling selective receptor modulators that act as agonists in some tissues and antagonists in others.
✅ Proven mechanisms that are frequently tested
GPCR Gs vs Gi: Gs stimulates adenylyl cyclase to raise cAMP and activate PKA, while Gi inhibits adenylyl cyclase, lowering cAMP; Gq activates PLCβ to produce IP3 and DAG, leading to Ca²⁺ release and PKC activation, with arrestin pathways providing parallel kinase signaling and desensitization.
RTK dimerization: Ligand cross‑linking stabilizes RTK dimers for trans‑autophosphorylation, switching on catalytic activity and assembling Ras–MAPK and PI3K–Akt signalosomes via SH2/PTB adaptors like Grb2 and Shc.
JAK–STAT triad: Cytokines bring receptor‑bound JAKs into proximity to trans‑activate, phosphorylating receptor tails and STATs; STAT dimers then drive transcription of genes governing survival, proliferation, and immune function.
Nuclear receptor transcription: Agonist‑bound receptors recruit LXXLL‑motif coactivators (e.g., p160, CBP/p300) with HAT activity to acetylate histone tails, relax chromatin, and facilitate RNA polymerase II–mediated transcription at response elements.
⚓ Practical study anchors
Remember the four classes—LGIC, GPCR, enzyme‑linked (RTK/JAK), and nuclear receptors—as the core scaffold for pharmacodynamics, linking drug targets to signaling speed, second messengers, and transcriptional outcomes.
Map exemplar pathways: LGIC→ion flux; GPCR→cAMP/IP3–DAG and β‑arrestin; RTK→MAPK/PI3K/PLC‑γ; JAK–STAT→direct gene regulation; nuclear receptor→chromatin‑level transcription—then attach drug classes and clinical time courses to each.
🏁 Bottom line
The pharmacology of drug action is concentrated at four receptor families – LGICs, GPCRs, enzyme-linked receptors and nuclear receptors – whose unique coupling mechanism determine differences in kinetics of signalling, clinical onset, efficacy, tolerance, and opportunities to pathway-selective therapy. Keeping these mechanisms linear is one of the keys to logically deduce and explain physiologic effects and AEs from the target to the drug in almost every therapeutic field.
Medical Disclaimer
The medical information on this post is for general educational purposes only and is provided by Pharmacology Mentor. While we strive to keep content current and accurate, Pharmacology Mentor makes no representations or warranties, express or implied, regarding the completeness, accuracy, reliability, suitability, or availability of the post, the website, or any information, products, services, or related graphics for any purpose. This content is not a substitute for professional medical advice, diagnosis, or treatment; always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition and never disregard or delay seeking professional advice because of something you have read here. Reliance on any information provided is solely at your own risk.
Community Notes
Join the Discussion
Ask follow-up questions, add clinical perspective, or share a useful clarification for the next reader.