


Introduction
Pharmacokinetics is the branch of pharmacology devoted to understanding what the body does to a drug once it has been administered. This entails a detailed look at four main processes collectively known by the acronym ADME: Absorption, Distribution, Metabolism, and Excretion. Together, these processes determine the onset, intensity, and duration of a drug’s action in the body (Goodman & Gilman, 2018).
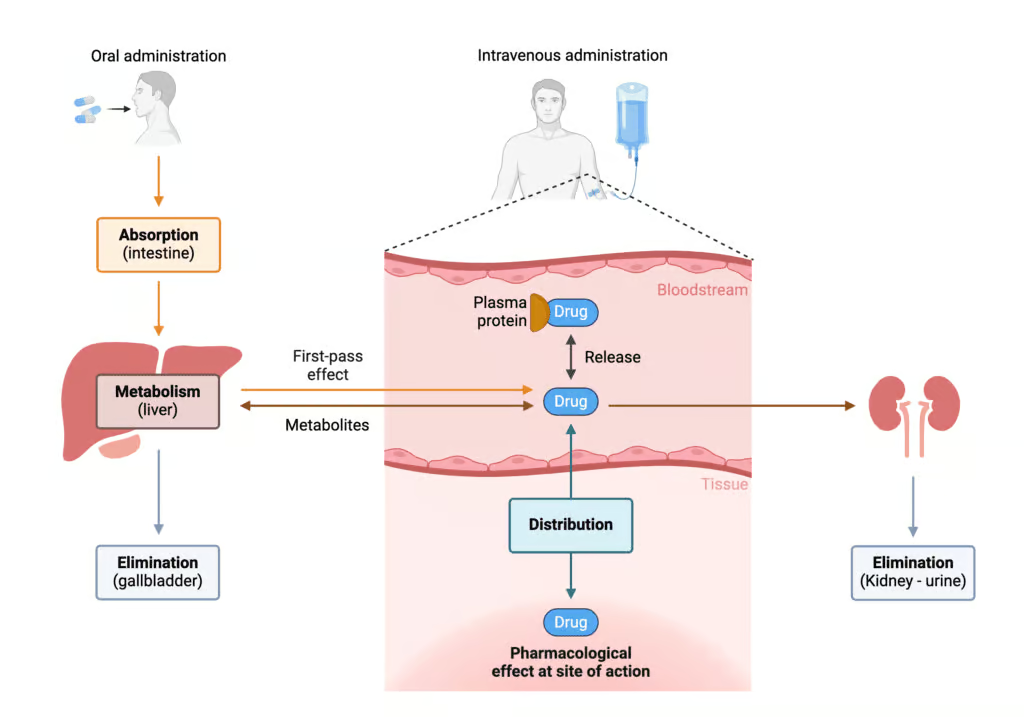
When a patient takes a medication—whether it is oral, injectable, topical, or inhalational—the substance must pass through numerous physiological barriers, enter circulation, distribute among tissues, undergo biochemical transformations, and eventually be eliminated in order to terminate its biologic effect. By understanding each of these four steps in detail, healthcare professionals and pharmaceutical scientists can optimize drug dosing and minimize adverse events (Katzung, 2020).
Within modern medicine, nearly all drugs—from acetaminophen for headaches to sophisticated biologics—undergo ADME processes, though to widely varying degrees. Subtle differences in, for instance, lipid solubility, molecular weight, protein binding, or enzyme susceptibility can profoundly shape a drug’s pharmacokinetic profile (Rang & Dale, 2019). The knowledge gleaned from pharmacokinetics is therefore crucial not just for clinicians deciding dosage regimens but also for drug developers attempting to engineer new therapeutic agents with ideal ADME characteristics.
This comprehensive overview provides an in-depth look at each of the ADME components, elaborating on the key factors that influence a drug’s journey through the body. By detailing common concepts—bioavailability, volume of distribution, hepatic biotransformation, renal clearance—and illustrating their practical implications with relevant examples, this discussion can serve as a valuable resource for physicians, pharmacists, researchers, and students seeking a more complete grasp of pharmacokinetic principles.
Absorption
Definition and Importance
Absorption refers to how a drug passes from its site of administration into the systemic circulation. For many common routes, such as oral (PO), intramuscular (IM), or subcutaneous (SC), the absorption phase is necessary to achieve therapeutic plasma concentrations of the drug (Katzung, 2020). The exceptions—intravenous (IV) or intra-arterial routes—bypass absorption because the drug is directly introduced into the bloodstream.
Achieving sufficient absorption is crucial, as too little means subtherapeutic responses, while excessive or too rapid absorption may cause toxicity. Bioavailability is the term denoting the fraction of an administered dose that actually reaches the systemic circulation in an unchanged form (Goodman & Gilman, 2018).
Factors Affecting Oral Absorption
- Physicochemical Properties of the Drug
- Lipid Solubility: Lipophilic compounds often penetrate membranes more efficiently, improving absorption. For example, diazepam (a benzodiazepine) is quite lipophilic, facilitating its ready absorption from the gut.
- Ionization State: Ionized molecules generally cross membranes less readily compared to non-ionized forms. Weak acids (e.g., aspirin) may be absorbed more in acidic environments, while weak bases (e.g., propranolol) can be better absorbed in slightly alkaline conditions.
- Molecular Size: Smaller molecules tend to diffuse more easily.
- Gastrointestinal (GI) Physiology
- pH and pKa: The GI environment changes from acidic in the stomach (pH ~1-3) to near-neutral or alkaline in the intestines (pH ~6-7.5). This gradient influences the proportion of drug in ionized versus non-ionized forms.
- Presence of Food: Food can slow gastric emptying, dilute the drug in the GI tract, or bind to it, thereby reducing absorption. For instance, tetracyclines chelate with calcium in dairy products.
- Motility and Transit Time: Enhanced motility (e.g., diarrhea) can limit a drug’s window of absorption, while slowed transit can extend drug contact time and partially boost absorption (Rang & Dale, 2019).
- Formulation and Delivery Systems
- Tablet Disintegration: If a tablet does not dissolve properly, absorption may be incomplete.
- Capsule Coatings: Enteric coatings shield certain proton-pump inhibitors or acid-labile drugs from the acidic stomach environment.
- Extended-Release (ER) vs. Immediate-Release (IR) formulations: ER products intentionally spread out drug absorption over a longer period, smoothing plasma concentration peaks (Katzung, 2020).
First-Pass Metabolism
A peculiar obstacle chiefly affecting oral drugs is first-pass or pre-systemic metabolism, primarily via the liver and sometimes through gut wall enzymes or intestinal microflora. For example, nitroglycerin is so extensively metabolized during its first pass through the liver that oral administration significantly diminishes its bioavailability, necessitating sublingual or transdermal routes (Goodman & Gilman, 2018).
Distribution
Definition and Overall Concept
Distribution is the process wherein a drug moves from the bloodstream into the interstitial and intracellular fluids of tissues to exert its pharmacological effect. The full distribution pattern depends on numerous factors, including organ blood flow, drug physicochemical properties, protein binding, and barriers (like the blood-brain barrier). After absorption (or injection), a drug rarely remains solely in the plasma; it partitions among various compartments such as muscle, fat, the brain, or others, typically seeking to establish equilibrium (Rang & Dale, 2019).
Volume of Distribution (Vd)
Volume of distribution (Vd) is a key parameter describing the relationship between the amount of drug in the body and its plasma concentration. Although it can be expressed in liters, Vd is often considered a “theoretical” volume that may not correspond to any real physiological space. For instance, a high Vd suggests extensive tissue penetration, while a low Vd indicates that the drug largely remains in the bloodstream (Goodman & Gilman, 2018).
- Low Vd (~3–5 L): The drug is restricted to plasma. Example: heparin often remains in the vascular compartment due to high plasma protein binding and large size.
- Moderate Vd (~12–20 L): Indicative of distribution within extracellular fluid but not extensively intracellular. Example: mannitol.
- High Vd (>40 L): The drug distributes widely in tissues, sometimes even accumulating in certain compartments like adipose tissue. Example: digoxin, with a Vd sometimes exceeding 400 L, reflecting high tissue affinity (Katzung, 2020).
Plasma Protein Binding
Certain drugs reversibly bind to albumin or other plasma proteins (e.g., α1-acid glycoprotein). While bound drug is inactive (cannot cross membranes or be metabolized effectively), free drug exerts pharmacological activity.
- Competition: Co-administration of two strongly protein-bound drugs (e.g., warfarin and sulfonamides) can lead to displacement, transiently increasing free plasma concentrations and clinical effects.
- Disease States: Hypoalbuminemia (e.g., resulting from liver disease) can elevate free drug fractions (Rang & Dale, 2019).
Special Barriers
- Blood-Brain Barrier (BBB): Tight junctions in brain capillaries allow only lipophilic or actively transported drugs—like levodopa—to cross effectively. Highly hydrophilic or large molecules (e.g., certain antibiotics) often have restricted entry to the CNS.
- Placental Transfer: Many drugs can traverse the placenta, but lipophilic, non-ionized drugs cross more readily. This underscores the need for caution when prescribing medication to pregnant women (Goodman & Gilman, 2018).
Metabolism
Purpose and Outcomes
Metabolism, often referred to as biotransformation, chemically modifies drug molecules to usually produce more polar, water-soluble derivatives (metabolites) that are more easily excreted. This process mainly occurs in the liver, though other sites—intestinal epithelium, lungs, kidneys—may also metabolize drugs (Katzung, 2020).In many cases, metabolism inactivates a drug. However, certain prodrugs (e.g., enalapril) depend on metabolic conversion to become active forms (e.g., enalaprilat), and sometimes metabolism yields toxic intermediates that can harm tissues if not further detoxified (Rang & Dale, 2019).
Phases of Metabolism
Metabolic reactions are conventionally categorized as Phase I (functionalization reactions) or Phase II (conjugation reactions). Some drugs undergo sequential Phase I then Phase II transformations, while others may only require one step.
- Phase I: Oxidation, Reduction, Hydrolysis
- Cytochrome P450 (CYP450) Enzymes: Vital for oxidative metabolism, particularly the CYP3A4, CYP2D6, CYP2C9, CYP1A2, and CYP2C19 isoforms. Many clinically relevant drug interactions stem from enzyme induction or inhibition. For instance, rifampin induces CYP3A4, accelerating metabolism of drugs like oral contraceptives, while ketoconazole inhibits CYP3A4, causing possible toxicity of co-administered drugs (Goodman & Gilman, 2018).
- Hydrolysis: Occurs with esterases (e.g., those acting on procaine) or amidases.
- Phase II: Conjugation
- Glucuronidation: Allows excretion of polar glucuronide conjugates. A typical example is morphine forming morphine-6-glucuronide, which retains analgesic activity.
- Sulfation: Creates sulfate conjugates, e.g., for acetaminophen.
- Acetylation: (e.g., isoniazid). Genetic polymorphisms in N-acetyltransferase can produce “slow” or “fast” acetylator phenotypes.
- Glutathione conjugation: Critical for detoxifying reactive intermediates, including the harmful metabolite of acetaminophen (NAPQI) (Rang & Dale, 2019).
Enzyme Induction and Inhibition
- Enzyme Induction: Repeated drug exposure (e.g., phenobarbital, carbamazepine) can upregulate CYP enzyme expression, enhancing metabolism and lowering plasma drug levels, sometimes causing therapeutic failure.
- Enzyme Inhibition: Drugs (e.g., cimetidine, fluoxetine) bind CYP enzymes, decreasing their metabolic capacity. Plasma levels of co-administered drugs may rise, risking toxicity.
Genetic Polymorphisms
Inter-individual variability in genes encoding metabolic enzymes can drastically alter drug handling. For instance, poor metabolizers of CYP2D6 may experience insufficient analgesia from codeine (since codeine requires metabolism to morphine) or unexpected toxicity if they have an ultra-rapid metabolizer phenotype (Katzung, 2020).
Clinical Relevance of Hepatic Metabolism
Patients with liver damage (e.g., cirrhosis, hepatitis) or compromised hepatic blood flow (e.g., in heart failure) may have reduced drug metabolism, leading to elevated risk of drug toxicity. Dosing adjustments become necessary for many medications, especially those with high hepatic extraction ratios (Goodman & Gilman, 2018).
Excretion
Overview
Excretion refers to how the drug and its metabolites are removed from the body. Renal excretion is the principal route for many drugs; hepatic excretion (including biliary pathways), pulmonary exhalation, and minor routes (sweat, saliva, breast milk) can also be significant. Eliminating the active compound is central for terminating pharmacological action, preventing accumulation and toxicity (Rang & Dale, 2019).
Renal Excretion
Understanding renal handling is critical, since the kidneys filter enormous volumes of plasma daily:
- Glomerular Filtration: Freely filters small, unbound drug molecules. Protein-bound drugs generally do not cross the filtration barrier. Glomerular filtration rate (GFR) influences how much drug is initially filtered.
- Tubular Secretion: Active transporters in the proximal tubule (e.g., OAT, OCT) can secrete acids/bases into the tubular fluid, facilitating removal. For instance, penicillin is rapidly cleared by active secretion.
- Tubular Reabsorption: Reabsorption of water can concentrate the drug in the tubules, encouraging passive diffusion back into the bloodstream if the drug is lipid-soluble or unionized under tubular pH conditions. Manipulating urine pH can help trap ionized drug in the urine—exploited clinically for overdose management (Katzung, 2020).
Hepatobiliary Excretion
The liver can excrete drugs into bile, which drains into the intestinal tract. Some conjugated metabolites (e.g., glucuronides) can undergo enterohepatic recycling if intestinal bacteria deconjugate them. This process may prolong a drug’s half-life. Estrogens found in oral contraceptives are partly recycled in this manner, which is why antibiotic therapy can reduce contraceptive efficacy (Goodman & Gilman, 2018).
Pulmonary and Other Minor Routes
- Exhalation: Volatile anesthetics, ethanol, or gaseous drugs largely exit via the lungs.
- Breast Milk: Drugs can diffuse into milk, posing risks to nursing infants. Lipid-soluble, basic drugs can accumulate. For instance, diazepam or codeine can appear in breast milk (Rang & Dale, 2019).
Clearance
Clearance (CL) quantifies the volume of plasma from which a substance is completely removed per unit time (e.g., mL/min or L/h). It encompasses all routes (renal, hepatic, etc.):
- Systemic Clearance = CL(hepatic) + CL(renal) + CL(others)
- A drug’s half-life (t½) is tied to clearance and volume of distribution according to t½ = (0.693 × Vd) / CL.
Drugs with high clearance vanish rapidly from plasma, requiring more frequent dosing or continuous infusion to maintain therapeutic levels. Aminoglycoside antibiotics, with relatively high clearance, are given at particular intervals to ensure efficacy while avoiding toxicity (Katzung, 2020).
Clinical Pharmacokinetics: Putting It All Together
Dose Regimens
Optimizing a dosing regimen typically strives to maintain plasma drug concentrations within a therapeutic window—above the minimum effective concentration but below the toxic threshold. By integrating data on bioavailability, volume of distribution, half-life, and clearance, clinicians can formulate loading and maintenance doses (Goodman & Gilman, 2018).
- Loading Dose: A larger initial dose to promptly achieve a desired plasma level. Formula: Loading Dose = (Vd × desired plasma concentration) / (bioavailability). For instance, digoxin often necessitates a loading dose because it has a large Vd and slow distribution phase.
- Maintenance Dose: Repeated or continuous doses replace drug lost through metabolism and excretion, stabilizing plasma concentration. Maintenance Dose = (CL × desired plasma concentration) / (bioavailability).
Multiple Dosing and Steady State
With repeated dosing at consistent time intervals (or via continuous IV infusion), steady-state is reached when the rate of drug administration equals the rate of elimination. Rule of Thumb: About 4–5 half-lives are needed to reach steady-state. This is why short half-life drugs (e.g., nitroglycerin with about 1–4 minutes IV half-life) equilibrate quickly, whereas a long half-life drug (e.g., amiodarone, 20–50 days) can take weeks or months to achieve steady-state (Rang & Dale, 2019).
Therapeutic Drug Monitoring
Some drugs possess a narrow therapeutic index—meaning the difference between efficacious levels and toxic concentrations is small. Examples: warfarin, digoxin, lithium, certain anticonvulsants. For these, measuring plasma drug levels is crucial, guiding dosage adjustment to maintain safe, effective concentrations. Pharmacokinetic modeling helps interpret these levels, factoring in patient variables like hepatic or renal function (Katzung, 2020).
Factors Influencing ADME Beyond Basic Physiology
Age
- Neonates and Infants: Immature hepatic enzymes, low GFR, and different body water composition can profoundly affect drug disposition. Dosing must be carefully adapted.
- Elderly: Declining renal function, potential hepatic changes, reduced total body water, and higher fat content can alter drug distribution and elimination. Polypharmacy poses further challenges due to increased drug-drug interactions (Goodman & Gilman, 2018).
Pathophysiological States
- Kidney Disease: Reduced GFR or tubular function hinders drug excretion. Dosages often require downward adjustments (e.g., for aminoglycosides).
- Liver Disease: Cirrhosis or hepatitis can reduce metabolic capacity, leading to drug accumulation. For instance, benzodiazepines or opioids might need lower or less frequent dosing.
- Heart Failure: Diminished blood flow to the liver or kidneys can hamper clearance, prolonging half-lives of certain medications (Katzung, 2020).
Pharmacogenetics
- CYP2D6 Polymorphisms: As noted earlier, these variants may cause a person to be a “poor,” “intermediate,” “extensive,” or “ultrarapid” metabolizer of various drugs.
- NAT2 Polymorphisms: Influence acetylation speed of isoniazid. Slow acetylators face increased risk of neuropathy; fast acetylators may require bigger doses.
- TPMT Variations: Affect mercaptopurine metabolism, crucial in leukemia chemo regimens.
Recognizing these differences can help tailor therapy to individual genetic profiles (Rang & Dale, 2019).
Practical Examples Illustrating ADME Concepts
- Acetaminophen
- Absorption: Rapid, near-complete from the GI tract.
- Distribution: Vd approximates total body water.
- Metabolism: Primarily hepatic conjugation (glucuronidation, sulfation). A small fraction forms a reactive intermediate (NAPQI), detoxified by glutathione. Overdose or depleted glutathione leads to hepatotoxicity.
- Excretion: Renal elimination of conjugates.
- Warfarin
- Absorption: High oral bioavailability, minimal first-pass metabolism.
- Distribution: Highly plasma protein-bound (~99%), small unbound fraction exerts effect.
- Metabolism: CYP2C9 is crucial in warfarin clearance. Polymorphisms or drug interactions with inducers/inhibitors can drastically shift INR.
- Excretion: Renal excretion of metabolites. Warfarin’s narrow therapeutic index demands careful monitoring (Goodman & Gilman, 2018).
- Beta-Lactam Antibiotics (e.g., Penicillin G)
- Absorption: Oral forms subject to partial inactivation by gastric acid.
- Distribution: Widely distributed into body fluids but not well in the CNS except when meninges are inflamed.
- Metabolism: Minimal hepatic metabolism.
- Excretion: Primarily renal tubular secretion. Probenecid competes for the same transporter, prolonging penicillin half-life (Rang & Dale, 2019).
- Digoxin
- Absorption: Variable from GI tract; some forms exhibit better bioavailability.
- Distribution: Large Vd, extensive distribution into muscle tissue.
- Metabolism: Relatively minimal hepatic metabolism. Some gut flora degrade it. Antibiotics can raise plasma digoxin levels by killing these bacteria.
- Excretion: Primarily renal clearance. Impaired renal function extends the half-life considerably (Katzung, 2020).
- Levothyroxine
- Absorption: Incomplete and can be influenced by food, antacids, or iron supplements.
- Distribution: Highly bound to plasma proteins (thyroxine-binding globulin).
- Metabolism: Hepatic deiodination, conversion to the more active T3 form.
- Excretion: Biliary excretion with some enterohepatic circulation. Owing to a long half-life (7 days), it requires several weeks to reach steady-state (Goodman & Gilman, 2018).
Optimizing Therapy Through ADME Knowledge
Dosing Adjustments in Renal Failure
When kidney function is compromised, drugs like aminoglycosides or vancomycin can accumulate. Monitoring creatinine clearance or estimated GFR guides the interval or dose amplitude modifications. This ensures adequate bacterial killing while mitigating nephrotoxicity or ototoxicity (Rang & Dale, 2019).
Impact of Enzyme Inducers or Inhibitors
Co-prescribing an enzyme inducer (e.g., rifampin) with oral contraceptives can undermine contraceptive effectiveness. Meanwhile, combining an enzyme inhibitor (e.g., fluconazole) with phenytoin can push phenytoin levels into the toxic range (Katzung, 2020).
Pediatric Dosage Formulations
Children’s metabolic pathways (particularly phase II reactions) continue to develop over months or years. Overexposure can easily occur if adult doses are inappropriately scaled down by weight alone. Strategies like population-based pediatric dosing guidelines or specialized formulations help ensure safe absorption, distribution, metabolism, and excretion in children (Goodman & Gilman, 2018).
Innovations in Drug Delivery
Advanced techniques—optimized nano-carriers, liposomal encapsulation, or prodrugs—seek to exploit ADME pathways for targeted drug release in particular tissues while minimizing systemic side effects. For instance, liposomal amphotericin B is less nephrotoxic because it is distributed differently than conventional forms, illustrating how adjusting distribution can profoundly affect toxicity profiles (Rang & Dale, 2019).
Conclusion
Pharmacokinetics, embodied in the ADME processes, forms the basis of understanding how drugs journey through the body. From the moment of administration to the final step of excretion, a cascade of intricate events—absorption across epithelial barriers, distribution into different tissues, metabolic transformations in the liver or elsewhere, and eventual clearance via renal or other routes—defines a drug’s therapeutic and toxicity profiles (Goodman & Gilman, 2018).
The practical relevance of these processes cannot be overstated. Prescribers must be cognizant of how variations in absorption might affect bioavailability, how distribution depends on protein binding and tissue solubility, and how metabolism can shape a drug’s half-life or produce toxic intermediates. Furthermore, excretion is the final determinant of drug accumulation; impairment in renal or hepatic function, or changes in enzymatic activity, can drastically raise the risk of adverse effects (Katzung, 2020).
As medicine continues to evolve, appreciating the nuances of ADME becomes increasingly paramount. Complex drug regimens in older adults, specialized pediatric dosing, genetically influenced enzyme variability, and advanced technologies for drug targeting all highlight the necessity for refined, patient-specific pharmacokinetic evaluations. By leveraging pharmacokinetic principles, healthcare professionals can tailor therapies to achieve efficacy, safety, and patient satisfaction, reinforcing the indispensable role of ADME knowledge in contemporary clinical practice (Rang & Dale, 2019).
References (Book Citations)
- Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 13th Edition.
- Katzung BG, Basic & Clinical Pharmacology, 14th Edition.
- Rang HP, Dale MM, Rang & Dale’s Pharmacology, 8th Edition.






